
Topographical features in the genome and how they’re helping us understand tumorigenesis
17 Aug 2023
This month, we launch the latest update to COSMIC mutational signatures which includes topographical analysis. In a special guest-edition to our blog, Dr Burcak Otlu talks us through the new data. Burcak is based at the University of California, San Diego and is an expert in the development of algorithmic solutions to emerging problems in genomics. Burcak Otlu is a member of the Cancer Grand Challenges Mutographs team funded by Cancer Research UK. Cancer Grand Challenges is a global funding initiative founded by Cancer Research UK and the National Cancer Institute in the US, seeking to tackle some of the toughest challenges in cancer research. As part of the Mutographs team, her work focuses on genome analysis with an emphasis on annotation and enrichment analysis of genomic loci and set operations on genomic datasets. Furthermore, she is interested in genomic variations, specifically, integrative analysis of variations in various cancer types. Keep reading to learn why topographical analysis is exposing processes behind tumorigenesis, how to interpret the graphs, and what she’s got planned next.

Why do we study topographical features?
DNA isn’t just a long string of base pairs with coding and non-coding regions. It can be thought of more like a long, folded winding molecular road – there are open and closed roads, shortcuts, tunnels, stop signs, pedestrian crossings, bridges, roadworks, and speed limits. Some roads are open only early in the morning and some are open late at night. Topography looks at the location and distribution of mutations in the genome with respect to these structural features and in terms of some special timings. Via topography analyses, we are focusing on (and these are explained more further down), open chromatin regions, nucleosome occupied regions, CTCF binding sites, post translational modifications at the histone tails, early and late replicating regions of the DNA, genic or intergenic regions of the DNA, leading and lagging strands, and transcribed, untranscribed and non-transcribed strands of DNA. In topography, we look at whether the mutations are enriched in these places, or they are depleted in these genomic regions of interest.
So, the answer as to why we study these topographical features is answered simply - because asymmetrical accumulation of mutations on the DNA strands and enrichments/depletions at specific regions of the genome may play roles in tumorigenesis and can lead to insights about the genomic basis of cancer.
Linking topographies with signatures
A new field of genetic enquiry is taking the world by storm. Mutational signatures identify unique patterns of mutations across the genome caused by specific endogenous and exogenous processes. These ‘molecular fingerprints’ are identified, verified, and stored on databases like COSMIC mutational signatures, but often we don’t understand the underlying causes.
Topographies can help to solve this. We study the location and proximity of mutations within each signature to topographical features. This allows us to understand if there are preferential distributions of mutations close to specific structural elements in the DNA, and thus whether a mutational signature is related to a specific topography. Such analysis can provide mechanistic insights about a mutational process, especially when the mutational signature has an unknown aetiology. For instance, if mutations are clustered in early replicating regions of the DNA, we can expect mis-regulated AID/APOBEC family of cytidine deaminases or faulty DNA-repair genes.
The methodology
The methodology behind topography is straightforward. Firstly, we download whole genome libraries which contain annotations of topographical features and their signals. The definition of ‘signal’ in this context is the likelihood that a DNA element is present at a particular location. If the signal is high, it means a high probability of a DNA element at that genome position, if it is low, it is less probable.
So, using these libraries, we annotate our whole genome samples taken from mutational signatures studies for topographical features, and measure the accumulation of signals for all mutations within a signature. The average signals are taken to represent the behaviour for that mutational signature.
This procedure is repeated for all mutational signatures in each cancer type, and also for all topographical features.
We also run topography analysis on simulation data rather than the real observed result. If we can get the same findings by simulated mutations, then our results are not something specific to the real mutations and therefore, not statistically significant. In other words, we look at simulated versus really real, to emulate the statistical significance of our findings.
It’s a long process, and you may wonder why we do it. The answer is that the topographical features can give us vital information regarding the underlying mechanisms of the mutational signatures.
Answering the unknowns
Let’s say we have a mutational signature with an unknown aetiology.
If we observe transcription standard bias then this mutational signature is most probably attributed to exogenous mutational process, including the ones due to environmental mutagens or even chemotherapy.
Extreme transcriptional strand bias and enrichment of mutations at the genic regions, instead of intergenic regions might imply that there is transcription coupled damage in this mutational signature.
If we observe replicational stand bias, most probably the signature is attributed to endogenous mutational process, which might be due to deficiencies in DNA polymerases (DNA proofreading enzymes) or aberrant activities of DNA enzymes.
Mutational signatures that exhibit both transcriptional and replication of strand bias, can be attributed to a mutational process due to defective DNA mismatch repair or deficiency in double strand break repair.
Looking at the case of tobacco smoke
To put this into context, SBS4 is a mutational signature linked to tobacco smoking. Our latest research has analysed the topography of SBS4 in five different cancer types. The mutagen responsible for SBS4 has the following distinctions:
- Mutations preferentially located in nucleosome occupied regions
- Have increasing mutation density from early to late replicating regions
- Causes transcriptional strand asymmetry with more mutations on the transcribed strands of the genome based on the pyrimidine base representation of the mutated base pair.
- Show depletions at some histone modifications.
Analysing the graphs
These findings can be better understood by looking at the graphs and their significance in-depth.
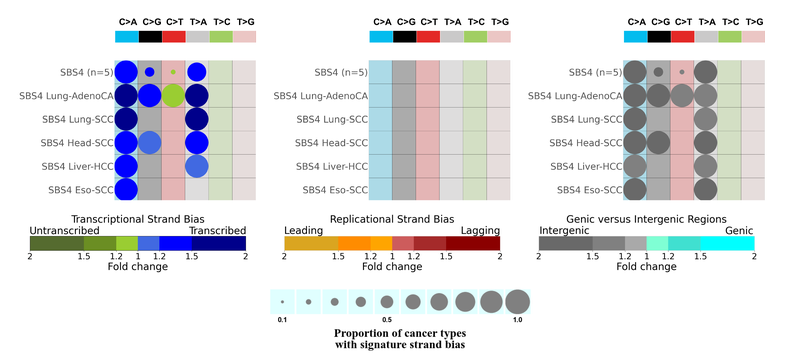
- Strand asymmetry
SBS4 mutations show transcriptional asymmetry with more mutations accumulated at the transcribed strand of the DNA with respect to the pyrimidine base representation, enriched at intergenic regions and no replicational strand asymmetry. And we also see that this is coherent across all five cancer types. We expect transcriptional strand bias and a lack of replicational strand bias for mutations caused by exogenous carcinogens.
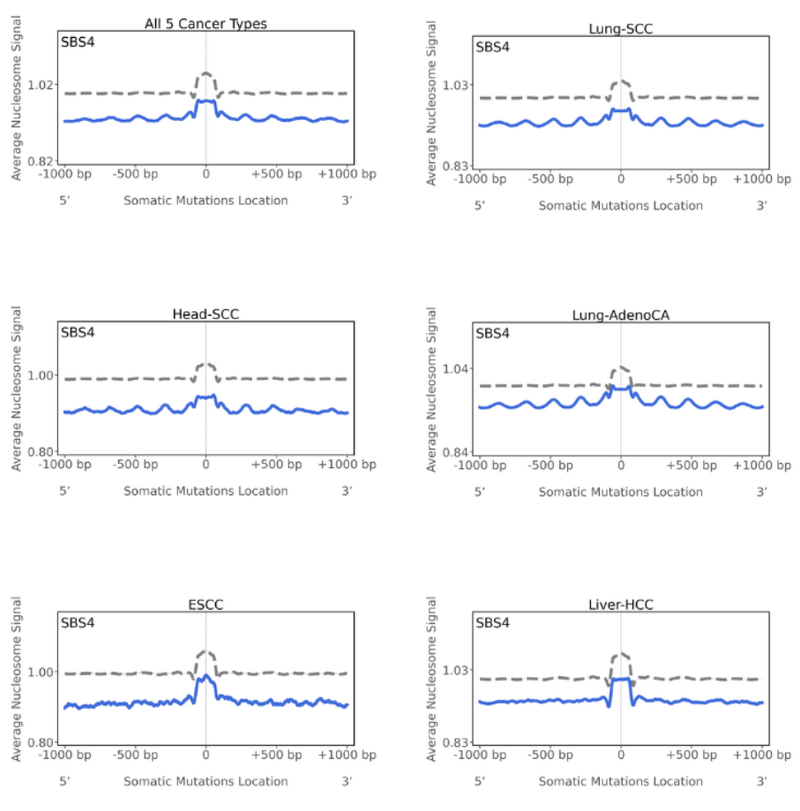
- Nucleosome occupancy
Nucleosomes are the basic packaging units of chromatin. They have four pairs of histone molecules and 147 bases wrap around these histone molecules with linker DNA in between. Nucleosomes are important in regulating gene expression, hence why they’re vital to study. Looking at the graph, a peak indicates that the mutations are preferentially located in the nucleosome occupied regions, whereas a trough would indicate the mutations are located within the linker-DNA or at regions with low average nucleosome signal. When we look at the SBS4 figures, we see that there is a peak at the mutations start position and this is the average behaviour for mutations across all five cancer types studied.
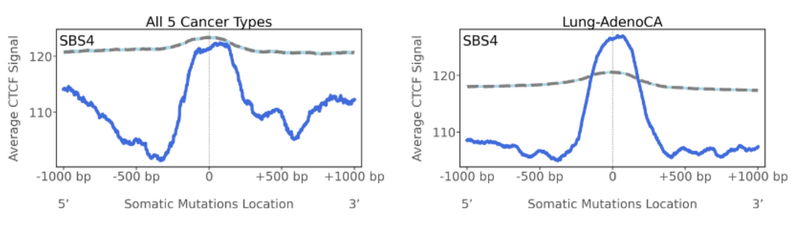
- CTCF
CTCF is a transcription factor and is referred to as the ‘master weaver’ of the genome. It’s a multi-purpose sequence specific DNA binding protein that has essential roles in transcriptional regulation, somatic recombination, and even chromatin architecture. The human genome harbours many CTCF binding sites, and it is those binding sites we look at as the topography feature here. Interestingly, for SBS4, we see a peak of mutations located at CTCF binding sites, but especially for lung-adenocarcinomas (in comparison to the straight line for the simulation). SBS4 signature in these cancers is likely disrupting the ability for CTCF to bind or creating de novo binding sites for other transcription factors. And this might have a role in the tumorigenesis and micro-environment of the cells.
- Early vs. late replicating regions
By this we’re referring to the density of mutations found at early replicated sites through to late replicated sites. Typically, highly expressed genes are found in the early replicated regions and have dedicated repair mechanisms in place to make sure these genes are protected. Deficiencies in DNA Repair pathways or excessive activities of AID/APOBEC family of cytidine deaminases results in more mutations in the early replicating regions. You can see this for SBS13, which is associated with AID/APOBEC activity. And this is common across many cancer types where we observe SBS13.
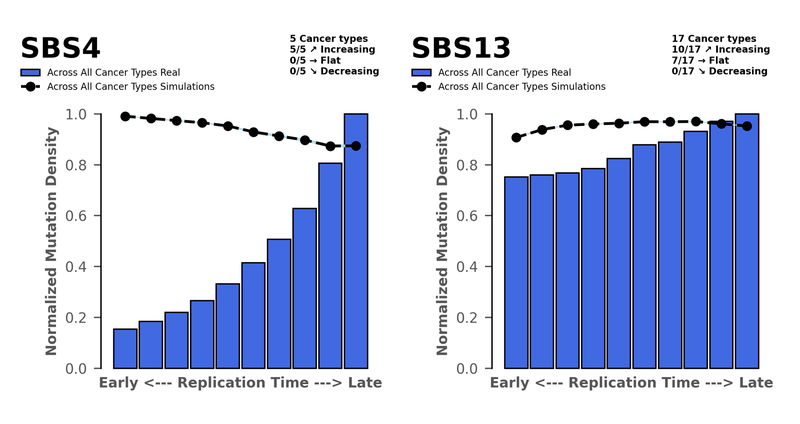
Whereas if the DNA repair pathways are conserved, they should be reversing much of the damage done by other mutagens. So, we only see mutations occurring at late replicating regions, where the editing isn’t as good. For SBS4 we see the highest density of mutations in the late sites, which is common across all five cancer types we studied. This is what we’d expect from an external mutagen such as tobacco smoke.
- Histone modifications
Relating mutational signatures with histone modifications known for active promoter, enhancer or repressor activities may provide further insights. For example, mutations enriched at intergenic, nucleosome depleted regions and strikingly clustered at active promoter and enhancer regions may indicate two things. Firstly, that existing transcription might be disrupted. Secondly, that de novo transcription binding sites might be created at these regulatory sites. Either (or both) of these could play a critical role in the tumorigenesis. Interaction network analysis between these regulatory regions and association with affected genes will be the further steps of our research.
We are mostly interested in histone post-translational modifications such as acetylation and methylation which are tightly involved in regulation of many cellular processes such as chromatin organization, DNA transcription, gene silencing, DNA replication and DNA repair. For example, histone acetylation results in loosened DNA and provides space for the transcription factors to come in and bind, enabling gene expression. Histone acetylation at H3 Lysine 27 and Lysine 9 (H3K27ac and H3K9ac) are associated with active enhancer and promoter regions, respectively. On the other hand, histone methylation at H3 Lysine 9 and Lysine 27 (H3K9me3 and H3K27me3) are both repressive marks by mediating heterochromatin formation and participating in gene silencing at euchromatin parts of the genome.
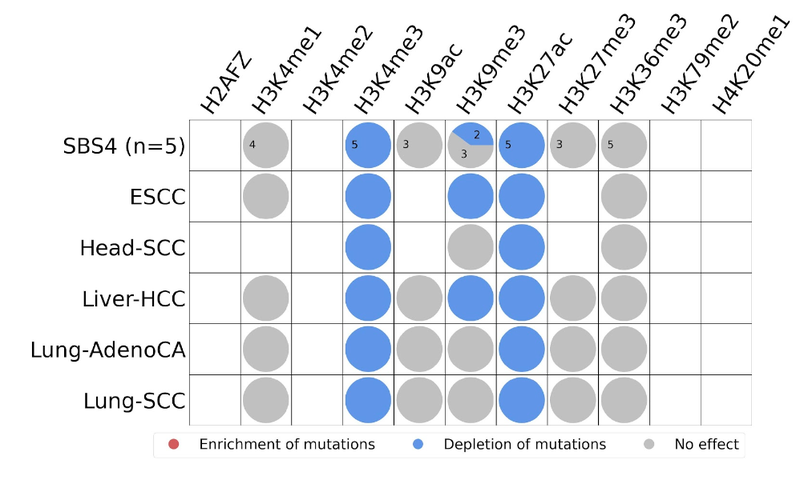
For our case study, we find that SBS4 mutations are either depleted or have no significant effect within these regions. Therefore, based on the overall average behaviour, they do not seem to have an important role in the tumorigenesis and this behaviour is consistent across five cancer types.
A bigger picture
Mutational signatures and topographical analysis are relatively in their infancy. But there is huge potential. In our latest research, we have analysed more than 70 mutational signatures across 40 cancer types. And we have provided this data on the COSMIC Mutational Signatures webpages. Our recently published paper also gives the important key findings across all mutational signatures and all cancer types coming from more than 5000 samples.
As to the future, I believe that looking at signatures through a topographical lens will enable us to identify key driver processes and mutations underlying cancer formation. At which point we create better diagnostic and prognostic tools. My next focus is to spend more time studying the mutations at transcription factor binding sites, active promoters, and enhancers. I want to know which genes have been affected by mutations at these sites and to unravel the interplay between non-coding regions and the coding regions and incorporate long-range genome wide chromatin interactions data.
- By Burcak Otlu