
Closing the care gap for rare cancers: Three examples in COSMIC
3 Feb 2022
Cancer was first described more than 4000 years ago, it wasn’t until the 20th century that we discovered alternative treatments to surgery, and chemotherapy wasn’t introduced until the 1940s. But now we’re in the midst of a genomics revolution and there’s an exponential growth in understanding. The more we learn, the bigger the challenge becomes, and it seems increasingly impossible to keep up to date.
For example, COSMIC has recorded more than 1500 unique cancer types to date.
For us, closing the care gap means being able to offer a precision medicine journey for each of them. The power to do this starts with pooling evidence from worldwide sources in one place to build a robust understanding of the molecular characteristics of each cancer. Then taking a step back to identify cross-cutting solutions based on molecular behaviours and genetic similarities between cancers rather than just their location.
It's a long journey to close the gap, but we've started. Explore a snapshot of how we're building knowledge for three rare-cancers in COSMIC below.
Rare lung cancer: Large cell neuroendocrine carcinoma (LCNEC)
Lung cancers can broadly be defined into two categories; Small cell lung cancers (SCLC) and non-small cell lung cancers (NSCLC). As the names suggest, they are differentiated by the size and shape of the cells that make up the tumours.
But this is a fairly reductionist way of thinking that doesn’t fit the bill for precision medicine. Let’s mix it up and discuss a rare lung cancer that straddles the categories.
Large cell neuroendocrine carcinoma (LCNEC) is a molecularly and biologically heterogeneous disease, sharing many molecular and histological features with small cell lung cancer and non-small cell lung cancer. LCNEC’s are aggressive with a poor prognosis and therapeutic management hasn’t changed for decades.
Evidence is emerging that some LCNEC’s evolve as a means of resistance to therapy. The normal evolution pathway in lung cancers goes from adenocarcinomas with EGFR mutations into NSCLC’s, with mutations which help them overcome treatment by tyrosine-kinase inhibitors. But both Kogo et al. and Zhao et al. have reported cases where the evolution was to a LCNEC instead. It’s yet to be determined why the two cases reported went ‘rogue’, but building evidence base suggests the involvement of of EGFR and RB1 - either through loss of expression or acquisition of further mutations.
To explain the significance of these genes, you can take a look at our Cancer Hallmarks Tool. But briefly, EGFR is a classic oncogene - the protein product finds a twin and buddies up to form a transmembrane receptor. Upon activation, this receptor kickstarts internal cellular processes such as angiogenesis and proliferation.

RB1 is a known tumour suppressor gene, and has a role in limiting the transcription of cell cycle genes. Studies to-date have implicated the Rb-protein in strengthening the innate immune response, regulation of cell death, and functioning as part of the network that responds to anti-mitogenic signals.
Another study brings clarity on LCNEC differentiation and relation to survival. It seems the location of the tumour is very important. Zhou et al. sub-classified LCNECs as centrally located (segmental and/or lobar bronchus invasion) or peripheral (without aforementioned invasion). They mapped the genetic profiles and overall survival of each subtype and found clear distinctions. Perhaps unsurprisingly, peripheral tumours were much more common and had a better survival, with an average of 4 years versus 1.5 years. Although this provides interesting prognostic information, the vital part for precision oncology lies in their distinct genomic profiles, namely in the two genes we’ve already referenced. First - EGFR mutations were only identified in samples from non-smokers with peripheral LCNEC. Secondly, the RB1 gene was more frequently expressed in peripheral tumours.
Which brings us onto an analysis which compared adenocarcinomas with LCNECs and provides hope for new treatment strategies. Whereas the case studies above indicated EGFR involvement in tumorigenesis, Makino et al. found no evidence of EGFR mutation in LCNEC’s. Instead, they displayed overexpression of topoisomerase II, somatostatin, and ERCC, which opens up the door to new targeted drug regimens.
Based upon this evidence, it’s unlikely that traditional platinum-based chemotherapy would be effective. Instead, agents such as etoposide or octreotide could be the heroes for patients with LCNECs. The former forms complexes with topoisomerase II – inducing double strand breaks, preventing repair and preventing replication, and eventually triggering cell death. The latter binds to somatostatin receptors – blocking downstream effects such as growth hormone activation.
Combined, these studies start to build a story of the importance of genetic profiling with rare tumours. Understanding the different drivers, histologies, and clinicopathologic features is important for ruling out some treatments and considering novel options. Most importantly, as seen above, this may be needed on a case-by-case basis rather than a one-size-fits-all approach.
Mesothelioma
The mesothelium is a layer of protective lining that covers the lungs (pleural lining), abdomen (peritoneal lining), heart (pericardial lining) and testes (tunica vaginalis). The primary function of the mesothelial cells that form this layer is to provide a slippery, non-adhesive, and protective barrier.
Malignant mesothelioma is a rare and aggressive tumour that affects this lining. The majority of cases occur in the pleura - known as Malignant Pleural Mesothelioma (MPM). In the UK, just under 3000 people will be diagnosed with mesothelioma each year, and only 7% of those are expected to survive five years or more. Mesothelioma is preventable in 94% of cases - but unfortunately it’s not a simple lifestyle change. This disease is (usually) caused by exposure to asbestos with a long lag time between exposure and presentation of disease. For instance MPM has an average latency period of 50 years. Since asbestos exposure is most common in industries with a male workforce, MPM is also seen predominantly in men.
In high income settings, policy changes around asbestos management, including training, identification, and safe removal mean cases of mesothelioma are decreasing. But the disease will remain a global health issue while asbestos continues to be mined and used, especially in developing countries.
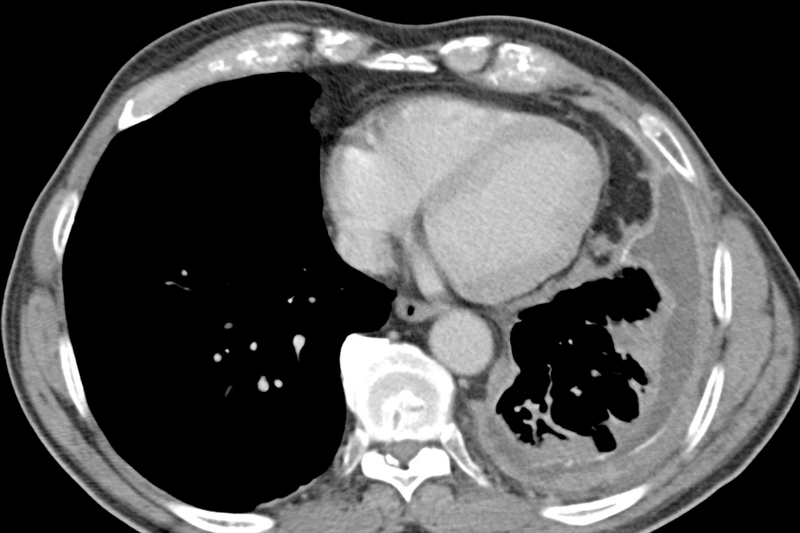
So what do we know about treatments?
If people present with early stage disease, surgery and radio-chemotherapy can be beneficial. But most patients are at an advanced stage when diagnosed. And traditional cytotoxic drugs are ineffective.
Because of this, a greater understanding of the underlying genetics of MPM and the development of novel targeted therapies are needed to improve the outcome for MPM patients.
COSMIC compiles that exact data. Here’s what we’ve included in our database.
An accumulation of studies including NGS whole exome sequencing have confirmed that the general genomic landscape of MPM includes frequent recurrent somatic mutations in the tumour suppressor genes, CDKN2A/B, NF2 and BAP1. There’s also evidence obtained through deep targeted sequencing that inactivating mutations in SETDB1 have a role to play.
SETDB1 codes for a histone methyltransferase which regulates histone methylation, gene silencing, and transcriptional repression. To be more precise, it specifically trimethylates 'Lys-9' of histone H3. SETDB1 is currently associated more closely with Huntingdon’s disease than cancer, but the strong indication of cancer involvement has placed it as a Tier 2 Census Gene in COSMIC’s database. If this evidence continues to grow, SETDB1 could present a novel option for epigenetic targeted treatments.
Moving from genomic profiling into translation to the clinic, there’s an emergence of distinct MPM subgroups which could benefit from targeted treatments. Tranchant et al. found an MPM molecular subgroup characterised by co-occurring mutations in the LATS2 and NF2 genes. Whereas Monch et al. identified a group of MPM characterised by overexpression of ALK.
In the former study, the authors defined the specific deregulated signal pathways in the subgroup and identified PF-04691502, an inhibitor of the mTOR/PI3K/AKT pathway, as potentially useful. Ongoing trials for PI3KA targeting drugs can be tracked via COSMIC Actionability.
Meanwhile, the latter study presented preclinical data showing that a combination of crizotinib (an ALK-1 inhibitor) and rapamycin may be suitable targeted therapy in MPM.
All of these studies show the power of genomic profiling in drug repurposing. Rapamycin is a classic example of this - originally developed as an anti-fungal agent, but repurposed to suppress the immune system, and then shown to be effective in cancers with specific mutations in the mTOR signalling pathways. Where overlap in mutational profiles occur across cancer (and other disease) types, a revolution can occur with the way trials are designed and drug-discovery pipelines are optimised.
Testicular Germ Cell Tumours
Testicular germ cell tumours (TGCT) are the most common testicular cancers and, although relatively rare, are the most frequent cancer type in younger men aged 15-49. Generally, the survival for this disease is very good, and the tumours respond excellently to combination cisplatin and etoposide based therapies. In the UK, more than 90% of patients will survive for 10 years or more.
Perhaps this is why the progress for understanding the molecular mechanisms for this disease seem to be relatively slow compared to other tumours. The current treatment protocol hasn’t changed for decades. It’s bereft of any molecular diagnostic or prognostic markers, and there are no targeted therapies. Granted, the old saying, ‘if it ain’t broke, don’t fix it’, comes to mind. But… What about those who don’t survive? And those who relapse? Approximately 20% will progress or relapse after first-line chemotherapy. How can we use molecular profiling to identify those who are most likely to relapse, or develop resistance? And how can we offer those people kinder treatment regimens or targeted therapies?
Answering those questions is why COSMIC gathers data on tumours with excellent survival as well as those with low survival.
Fortunately, advances in next generation sequencing have enabled the genomic landscape of TGCT to be better studied. And an interesting picture is being built up. Firstly, recurrent somatic mutations in KIT, KRAS, BRAF and NRAS are reported in TGCT. Secondly, point mutations are fairly uncommon and TP53 is rarely mutated. Thirdly, WT1 has been identified as a new gene involved in TGCT pathogenesis and could be useful as a prognostic marker. WT1’s involvement should perhaps be expected, as it encodes a transcription factor which is essential for testis development and function.
Thinking back to our original questions, and what we can actually do for those with resistant or relapsed TGCT, a recent study from Ross et al. provides some answers. Genetic profiling of those with chemorefractory TGCT highlighted a high frequency of KRAS genomic alterations. As the KRAS-pathway is commonly mutated across cancer types, targeted therapies have already been developed and trials with these agents could be considered for resistant/relapsed TGCT too.
The study’s aim was to look specifically at the similarities/differences in mutational profiles between two subtypes of TGCT which had become chemo-refractory; seminomas and nonseminomas. With the exception of KRAS, they found that the two subtypes had distinctly different genomic profiles. Non-seminoma TGCTs displayed a high tumour mutational burden and microsatellite instability compared to the seminomas. In tumours with high mutational burden, immunotherapies tend to be more effective, so this could represent an option for patients who display this profile.
The challenge for these tumours comes in the high survival. With the average drug development pathway taking billions of pounds and more than a decade - can we justify the health economics of developing a novel targeted drug for a cancer where the majority survive? Maybe not. But we certainly could justify designing trials with inclusion criteria based on mutational profiles rather than cancer types. Hence the ongoing need to record and accumulate the data in a database that covers all cancers.
Take home message: Closing the care gap through an unparalleled somatic mutation resource
It can be difficult to infer causation, design new treatments, or justify health economics when it comes to rare cancers. Scientific design is typically based upon statistically significant results based upon representative samples, so a single case study or handful of cases doesn’t provide much hope. This is where COSMIC is stepping up.
We're closing the care gap by accumulating data. We enable users to explore our database from a gene-centric view, a cancer-subtype view, specific mutational significance, or a look through existing treatments and ongoing trials. Combined, our products paint a genomic picture that’s unparallelled, and enables us to be the trusted foundation for cancer precision-oncology research.